

【摘要】目的 :代谢相关脂肪肝( MAFLD )的发病机制复杂。脂质代谢紊乱、慢性炎症和氧化应激是 MAFLD 的核心事件。饮食干预是预防 MAFLD 发病和进展的重要治疗策略。丁酸梭菌( CB )和可溶性膳食纤维( SDF )通常被认为对健康有益。我们探讨了两种针对微生物群的干预措施( SDF 和 CB )如何影响 MAFLD 小鼠的肝脏免疫系统、氧化应激和脂质代谢。方法:通过给 C57BL/6 小鼠喂食高脂饮食( HFD ),建立 MAFLD 小鼠模型,探讨SDF 和 CB 在 MAFLD 中的作用。干预 8 周后,我们测量了小鼠肝脏的免疫细胞功能、脂质代谢和氧化应激水平。结果:单次 SDF 或 CB 干预对改善 MAFLD 无效;然而,与 SDF 和 CB 的联合干预增加了微生物多样性,减少了炎症、氧化应激和脂质合成。此外,我们确定,通过激活 Acly/Nrf2/NF-κ B 信号通路,与 SDF 和 CB 共同干预介导的脂肪酸氧化。最重要的是,联合干预通过抑制巨噬细胞分化为促炎性 M1 巨噬细胞发挥抗炎作用。结论:本研究表明, SDF 和 CB 的联合干预可以改善 MAFLD , SDF 和 CB 的联合干预可能是 MAFLD 潜在的肠道微生物群调节剂和治疗物质。
【介绍】
过去20年的大量研究清楚地表明,非酒精性脂肪肝(N A FLD )是一种代谢性疾病。与全身代谢功能障碍相关的脂肪肝很常见,影响全球约24%的成年人。在这种高发病率的背景下,专家们达成了共识,即非酒精性脂肪肝并不反映当前的知识,以及代谢相关疾病的新概念脂肪性肝病“MAFLD ”的提出是为了克服MAFLD定义的局限性。 MAFLD 代表从单纯性脂肪变性到非酒精性脂肪性肝炎(NASH )的一系列疾病状态,包括纤维化、肝硬化和肝细胞癌。随着饮食和生活方式的改变,MAFLD 已发展成为一个全球健康问题。然而,MAFLD 的发病机制尚不清楚。它是一种复杂的多因素疾病,涉及脂质代谢、线粒体功能障碍、肠道菌群紊乱、巨噬细胞编程和表观遗传改变,这些改变了肝脏微环境,会倾向于肝脏损伤。因此,MAFLD的特点是慢性炎症、简单脂肪变性和低水平的氧化应激反应。肝脂肪变性、炎症、内毒素和氧化应激之间的相互作用已得到充分描述。大量肝脏脂肪堆积导致促炎因子(TNF-α、IL-6和IL-1β)释放,并导致促炎环境。此外,巨噬细胞是肝脏炎症的关键影响细胞。肝脏中M1和M2巨噬细胞的极化是炎症反应、肝细胞损伤和活化的重要步骤-肝卫星细胞的分化。在M AFLD 患者中,常伴有肠道微生物群失调和肠道粘膜屏障功能受损,使脂多糖(LPS)易于通过门静脉到达肝脏。值得注意的是,M1巨噬细胞被LPS、TNF-α和IFN -γ激活。M1巨噬细胞主要参与核因子κB(NF-κB)激活,并产生大量活性氧(ROS)以促进氧化应激。在肝脏中,氧化应激和炎症导致肝脏脂肪变性。综上所述,随着MAFLD 的进展,炎症、氧化应激、内毒素和脂质沉积相互促进,形成正反馈回路。因此,改善肝脏脂肪变性、氧化应激和肝脏炎症是治疗MAFLD的一种有前途的策略。目前,除了改变生活方式,包括饮食和锻炼外,没有批准的MAFLD 药物治疗。可溶性膳食纤维(SD F)和益生菌是通过调节肠道微生物群来调节脂质代谢和炎症的相对简单的方法。丁酸梭菌(CB)通过保护肠道粘膜屏障完整性和减少炎症因子改善结肠炎。SDF促进脂质代谢,并降低体重和血糖水平。此外,SDF的CB发酵会产生短链脂肪酸(丁酸、丙酸和醋酸盐),这是肠道内稳态的重要决定因素,与宿主脂质代谢密切相关。然而,CB和SDF在MAFLD 中肝脏脂肪变性、氧化应激和肝脏炎症相互作用中的作用尚不清楚。
【材料和方法】
2.1 动物实验
所有实验程序均按照国立卫生研究院指南进行,并经浙江大 学 动 物 伦 理 委 员 会 批 准 。 雄 性 C57BL/6小 鼠(18-20g),约6-8周大,购自中国上海斯莱克公司。给小鼠喂食正常的周饮食([NCD ],1025,脂肪含量4%卡路里;HFKbio,中国)或高脂肪饮食([HFD ],D 12492,脂肪含量60%卡路里;研究饮食,中国)24周。有三个不同的治疗组:SDF组(HFD -SD F)、CB组(HFD -CB)和SDF和CB同时干预组(HFD -SC)。对于治疗组,CB(剂量为2×109 CFU /0.2 m L)26与H FD 喂养同时给予,每周3次,持续8周(H FD 喂养24周后)。HFD -SD F组和HFD -SC 组接受含有SDF(7g/L)的饮用水 ),同时,NCD -PBS组和HFD -PBS组每周3次灌胃0.6 m L无菌PBS,持续8周(图1A )。SDF(7克升-1)购自北京同泽康成医疗科技有限公司(成分:低聚半乳糖、水苏糖、甘露糖寡糖)。27CB(ATCC19398)购自中国微生物总培养收集中心。
2.2 细胞和酪酸梭菌的培养
L02细胞在含有10%胎牛血清(Therm o Fisher Scientific,Inc.)和1%抗生素-抗真菌剂的DMEM (Therm o FisherScientific,Inc.)中培养。将约5×105个细胞接种到12孔板中(每组3孔)。用15μg m L处理L02细胞1棕榈酸(PA)48小时,作为MAFLD 模型细胞。在这48小时内,干预组接受不同浓度的丁酸(200、400和600μM )治疗。CB在37°C的厌氧条件下在脑心输液培养基中培养24小时,直到在A600的细菌密度为0.5的对数生长期。通过离心(3000g×5分钟)收集细菌,并在无菌磷酸盐缓冲液(PBS)中重新悬浮至最终实验浓度2×109 CFU /0.2 m L,该浓度是根据之前的研究和初步实验选择的。26收集上清液,通过0.22μm 孔径的过滤器过滤,然后用完整的培养基稀释。
2.3 组织学分析
将肝组织在多聚甲醛(4%)中固定24小时,并将其包埋在PA RAffin中。对于苏木精和曙红(h&E)和M asson三色染色,将PARA ffin包埋的样品分别切片为5µm 和3µm 厚。使用Masson的三色染色试剂盒(Solarbio,G 1340,中国)进行Masson三色染色。使用苏木精-伊红/HE染色试剂盒(Solarbio,G1120,中国)进行H&E染色。然后使用点滑扫描系统(英国海上索森德奥林巴斯)观察组织切片。
2.4 蛋白质印迹分析
使用添加蛋白酶抑制剂和蛋白酶抑制剂的RIPA buffer提取组织和细胞蛋白质。用适当的抗核因子红系-2相关因子2(Nrf2)(12721,细胞信号;稀释度:1:1000)、Keap1(98047,细胞信号;稀释度:1:1000)、HO-1(822069,细胞信号 ;稀释度1:1000)、 toll样 受 体 4(TLR4)(14358,细胞信号;稀释度:1:1000)、半胱天冬酶-1(Ab179515,Abcam ;稀释度:1:1000)的一级抗体孵育细胞膜,IL-1β(#27989,细胞信号;稀释度:1:1000)和脂肪酸和脂质代谢抗体(#8335,细胞信号;稀释度:1:1000)。β-肌动蛋白(#AF0003,Beyotim e;稀释度:1:1000)和GAPDH (#1E6D 9,Proteintech;稀释度:1:1000)用作蛋白质的内部对照。使用Im ageJ软件通过密度测定法对蛋白质进行定量。
2.5 序列分析
16S rRNA基因测序由诺沃金生物信息技术有限公司(中国北京)进行。使用天根试剂盒从冷冻粪便样本中分离细菌DNA 。通过PCR扩增16S rRNA基因的V3–V4高变区。测序在IlluminaHiseq平台上进行(Illum ina,圣地亚哥,加利福尼亚州,美国)。这个
QIIME平台用于将读取数据聚类到操作分类单元(OTU )、质量控制和微生物数据分析中。转录组测序由诺沃金生物信息技术有限公司(中国北京)进行。测序库是使用NEBN ext®Ultra构建的™ 按照制造商的说明,定向RNA库制备试剂盒(美国NEB)。GO seqR包用于差异表达基因的GO富集分析。KEGG被用来从分子水平上理解生物系统的功能。
2.6 流式细胞术
将各组的肝组织均质,制备单细胞悬液。用以下抗小鼠抗体 对单个肝细胞进行染色:抗CD45-FITC (克隆30-F11; BioLeg end,美国 )、 抗CD11b-APC(克隆M1/70,BioLeg end)、抗F480 -BV421(克隆BM8;BioLegend,美国)、抗CD206-PE(克隆C068C2;BioLegend,美国)和抗MHCII-PECy7(克隆M5/114;BioLegend,美国)。使用 FV S78 0(BD Biosciences,U SA )测定巨噬细胞的数量。使 用BD 进行流式细胞术™ LSRFortessa公司 (BD Biosciences)。使用FlowJo软件v10.0(BDBiosciences)进行分析。根据制造商的说明,使用带有V-底板V02(目录号740379;BioLeg end,美国)的小鼠面板 (5-Plex)对血浆细胞因子进行定量。分析的细胞因子包括:IFN-γ、TNF-α、IL-6、IL-10和IL-1β。
2.7 酶联免疫吸附试验(ELISA)
称重肝组织(30mg)并在PBS中均质,以制备10%的匀浆。使用ELISA试剂盒(L21123317,Uscnk,中国武汉)测定肝匀浆中的LPS水平。将标准品和样品在室温下镀在预先涂有LPS特异性抗体的微孔板上2小时。孵育后,清洗样品,然后用HRP结合的抗小鼠IgG(1:1000)孵育。洗涤后,在450nm处读取标准品和样品。数值以pg-ml表示1.
2.8 统计分析
使用SPSS 21.0进行统计分析。测量数据以平均值±标准差表示。使用双向ana分析各组间的比较-方差分析(ANOVA)和最小显著差异(LSD)。所有P值均为双尾,P值<0.05的差异具有统计学意义。
【后果】
3.1 SDF和CB的联合干预改善了HFD诱导的MAFLD
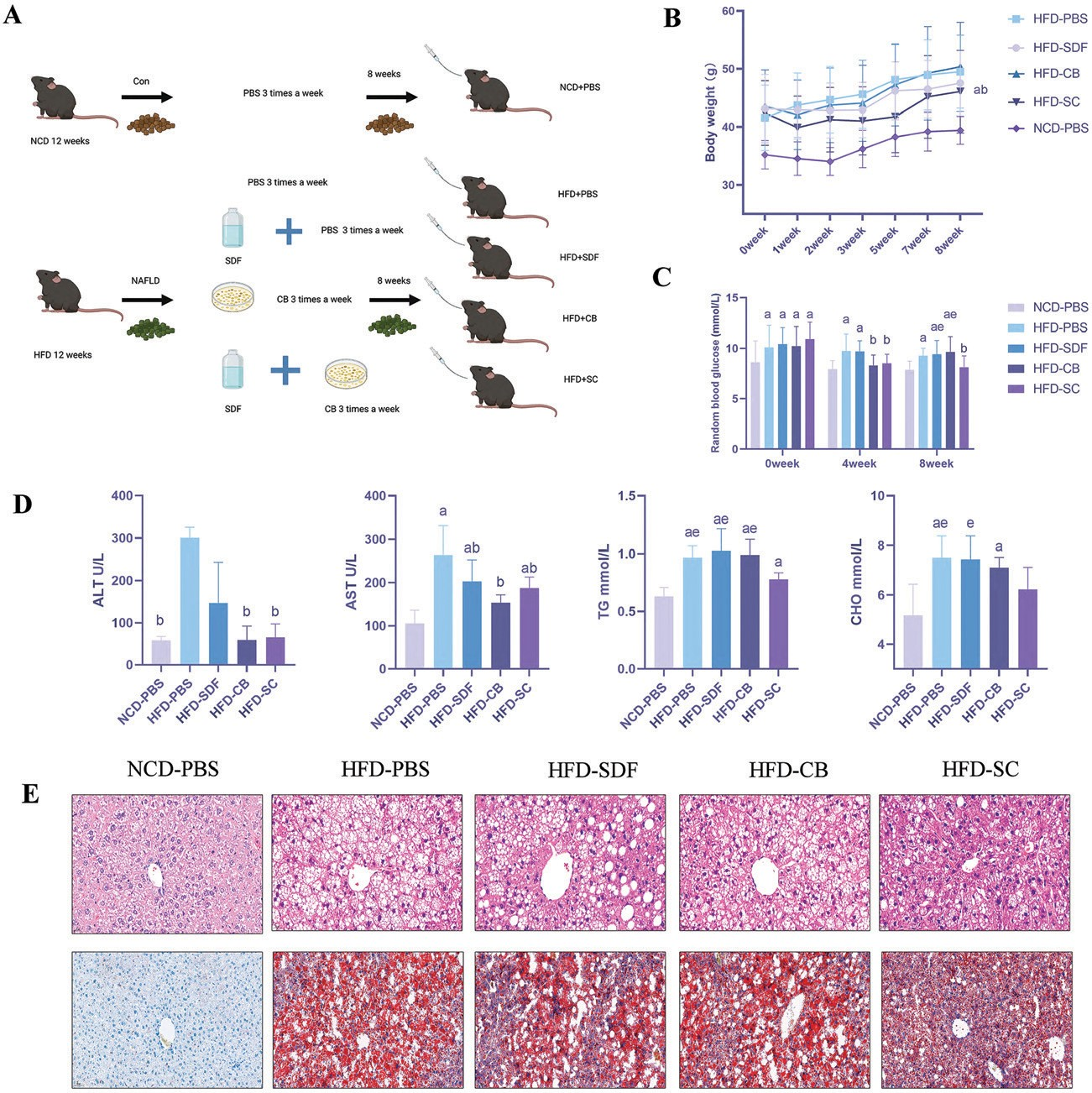
实验期间的重量变化如图1B所示。饮食干预前,HFD喂养的小鼠体重显著高于NCD喂养的小鼠,表明成功建立了MAFLD模型。干预8周后,HFD-SC组的体重显著低于HFD-PBS组(p<0.05)。然而,在H FD -PBS小鼠和两个 单一干预组之间未观察到体重的显著变化。此外,我们在第一个月没有检测到四个HFD喂养组(HFD-PBS、HFD-CB、HFD-SDF和HFD-SC)之间的随机血糖差异(图1C )。与HFD-PBS组相比,HFD-SC 组在第4周和第8周的随机血糖水平显著降低(p<0.05)。值得注意的是,HFD-CB组在第4周的血糖水平低于HFD-PBS组,但这一现象在第8周消失。我们认为这与长期接触HFD有关。与HFD-PBS组相比,HFD-SC组的肝丙氨酸转氨酶(ALT)、天冬氨酸转氨酶(ASPA)显著降低-胃酸转氨酶(AST)、甘油三酯(TG)和胆固醇(CHO)水平(p<0.05;图1D)。肝切片用H&E和油红O染色,以证明不同处理对MAFLD进展的影响(图1E)。H&E和油红0染色结果显示,NCD喂养小鼠的肝组织完整,细胞清晰可见,肝小叶结构完整,没有脂质积聚。相反,HFD喂养的小鼠表现出松散和伸展的肝细胞、肝细胞气球样变、小叶炎症和小叶脂质填充的细胞内空泡。此外,在四组HFD喂养的小鼠中,与HFD-PBS组相比,HFD-SC组观察到的上述病理变化有所改善。生化检查和肝组织病理学数据显示,SDF和CB联合干预可显著减轻HFD-PBS小鼠的肝脂肪变性。
3.2 SDF和CB联合干预改善HFD 诱导的炎症
慢性炎症是MAFLD的一个重要特征。值得注意的是,MAFLD的特征是肝脏中单核细胞源性巨噬细胞(MOMF)的增加。MOMF和Kupffer细胞(KC)分泌一系列促炎细胞因子,引起肝脏和全身炎症。因此,我们检测了肝脏MOMF和血清炎症因子水平(TNF-α、IFN-γ、IL-1β、IL-10和IL-6)。与对照组相比,HFD -PBS组的肝脏KC百分比增加(图2A和图2)
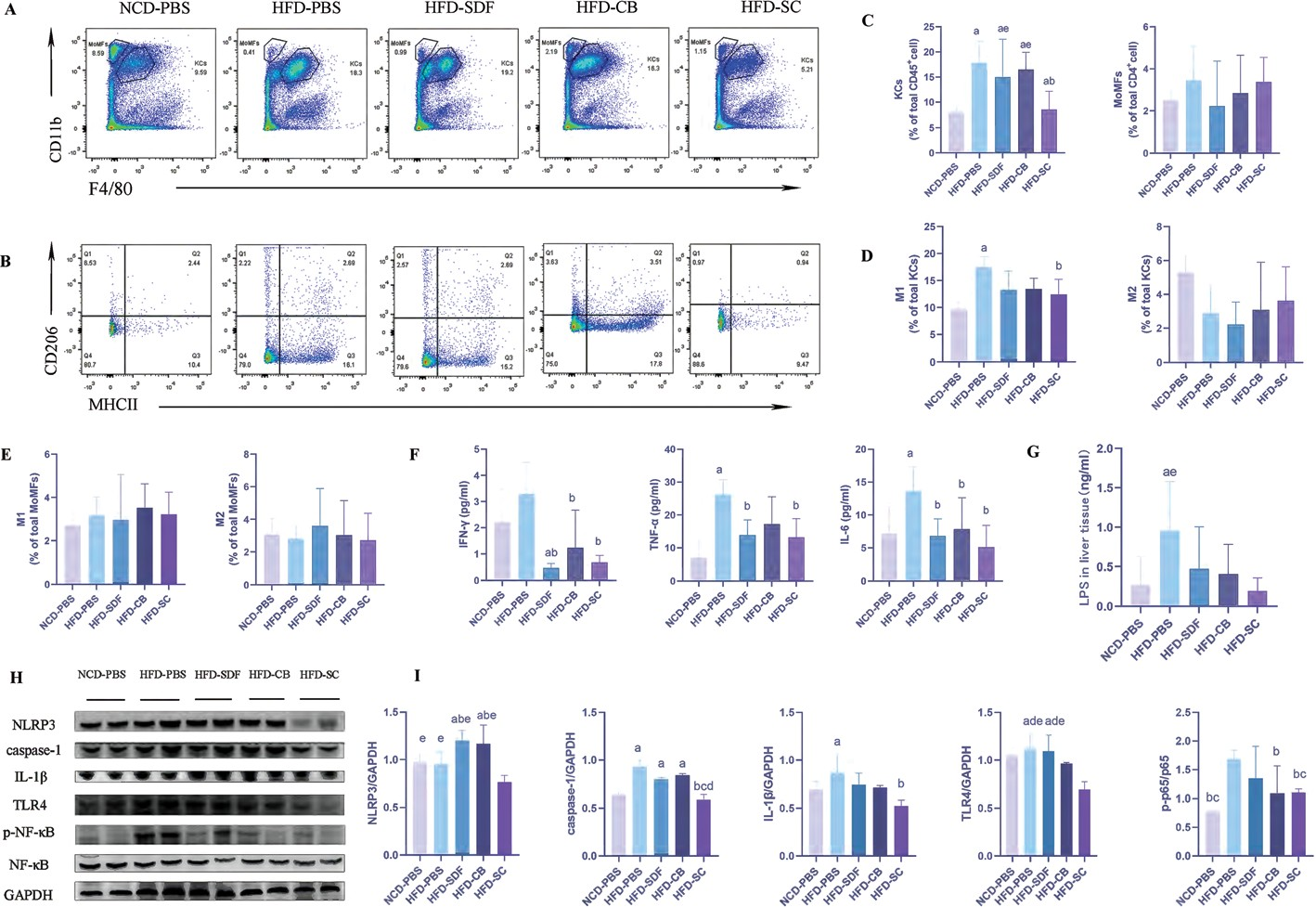
图2 SDF 和 CB 联合干预抑制 HFD 诱导的炎症。( A )肝内巨噬细胞( KC 和 MOMF )的代表性流式细胞术图。( B )KC中M1/M2巨噬细胞的代表性流式细胞术图。( C )肝CD45+细胞中巨噬细胞的百分比。( D ) KC中M1/M2巨噬细胞的极化。( E )MOMF 中 M1/M2 巨噬细胞的极化。( F )血清炎症因子水平。( G )肝脏中NLRP3和p-P65的代表性免疫印迹分析。( H )NLRP3和p-P65丰度的定量分析。数据以平均值±标准差表示,每组n=4–7 。a:p<0.05 ;与 NCD-PBS 相比, b:p<0.05 ;与 HFD-PBS 相比;c:p<0.05 ;与HFD-CB相比;d:p<0.05 ;与HFD-SDF相比;e:p<0.05;与HFD-SC相比。KC极化为M1促炎表型(图2B和D )。此外,与SDF和CB的联合干预显著降低了肝脏KC的百分比,并抑制了M 1巨噬细胞的激活。然而,单用SD F或CB干预对MOMF和M1巨噬细胞没有类似的影响。M 2巨噬细胞的百分比在各组之间没有差异(图2E)。同时,H FD -PBS组的血清TN F-α、IFN -γ和IL-6水平也升高。与上述结果一致,H FD -SC组中上述三种炎性细胞因子的水平低于HFD-PBS组(图2F)。然而,各组中IL-1β和IL-10的浓度过低,无法检测到。上述结果表明,与SD F和CB的联合干预通过介导KC分化改善了M AFLD 小鼠的炎症反应。膳食饱和脂肪酸诱导的NF-κB激活和核移位导致产生促炎 症细胞因子 (IL-6、 TN F-α和 IL-1β)。31,32N F-κB信号通路也通过介导M1巨噬细胞的激活而成为MAFLD的主要贡献者。此外,LPS刺激M1巨噬细胞激活NF-κB,从而诱导NOD 样受体家族3(N LRP3)的表达。考虑炎症和MAFLD之间的关系,以及NF-κB对MAFLD的调节作用M1巨噬细胞激活,进行western印迹以确定N LRP3炎症体(NLRP3、IL-1β和caspase-1)和TLR4/NF-κB信号通路的激活(图2H 和I)。此外,我们还检测了肝组织匀浆中LPS的水平。正如预期的那样,HFD治疗显著增加了LPS的水平,并通过TLR4诱导NF-κB的磷酸化。此外,与SDF和CB联合干预可有效降低LPS水平 (图2G ),并抑制TLR4/N F-κB信号通路的激活。一直以来,与SD F和CB的联合干预对N LRP3炎症体的激活产生了类似的影响。
3.3 在HFD 诱导的MAFLD 中,SDF和CB改变了基因转录
为了确定SDF和CB联合干预如何改善MAFLD,使用转录组测序分析对不同组的肝组织转录组进行测序。各组之间的基因表达差异如图3A所示。分别分析了不同组中上调和下调的基因。基于层次聚类分析,NCD-PBS、HFD-PBS和HFD-SC组分别显示出显著的聚类趋势(图3B),而HFD-CB和HFD-SDF组没有显示出显著的聚类。差异基因通过GO 富集和KEG G 途径分析进行处理。图中显示了KEGG富集程度最高的前20项。与HFD-PBS组相比,KEGG富集分析表明,在H FD -CB组中,主要富集物为“紧密连接”(图3C)。此外,在HFD-SDF组中,GO主要富集物为“脂质生物合成过程”、“类固醇生物合成/代谢过程”和“乙酰辅酶A/acly辅酶A代谢过程”(图3D )。GO分析表明,这些基因主要富集于抗氧化应激途径(“NADH脱氢酶(醌/泛醌)活性”、“氧化还原酶复合物/活性”、“过氧化物酶活性”,以及HFD-PBS和HFD-SC组之间的“抗氧化活性”)和线粒体功能(“ATP合成/代谢过程”、“线粒体呼吸链”、“线粒体内膜”和“线粒体蛋白质复合体”)(图3E)。KEGG途径的主要富集物是H FD -PBS组和H FD -SC组之间的“谷胱甘肽代谢”、“脂肪酸降解”、“MAFLD”和与抗氧化障碍相关的疾病(“阿尔茨海默病”和“帕金森病”)以及“M AFLD ”(图3F)。因此,我们推测SDF和/或C B通过调节肠道微生物群结构影响肠道紧密连接、脂质代谢和抗氧化应激功能。
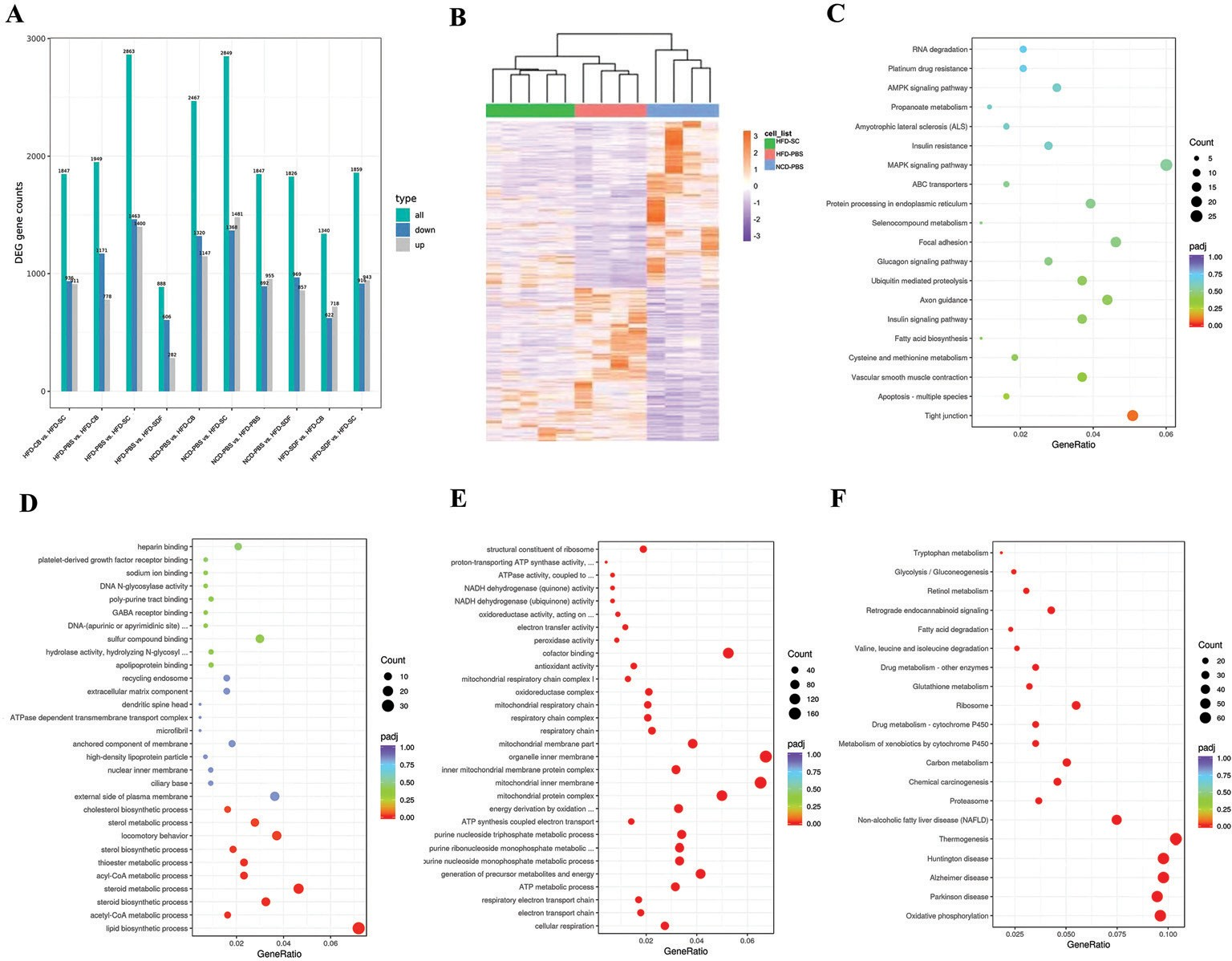
图3 SDF和CB对肝脏基因转录的影响。( A )不同组合中差异表达基因的数量。( B )差异表达基因的聚类热图。( C ) HFD-PBS组和HFD-CB组之间的富集KEGG项散点图。( D ) HFD-PBS组和HFD-SDF组之间的富集GO项散点图。( E ) HFD-PBS组和HFD-SC组之间的富集GO项散点图。( F )HFD-PBS组和HFD-SC组之间的富集KEGG项散点图。 n=4-5/组。
3.4 SDF和CB联合干预改善HFD诱导的肠道微生物群失调
为了确定改变肠道微生物群的组成和结构是否可以改善脂质代谢,先天免疫系统、氧化应激和肠道粘膜屏障的组成,以及为了探索干预是否在肠道微生物群或其生物学方面产生了特征性变化,在不同干预组中评估了微生物群组成、多样性和功能的变化。通过Simpson、Shannon和C hao1指数评估肠道微生物群的α多 样性。与NCD-PBS组相比,我们观察到HFD-PBS组的α多样性指数显著降低(p<0.05;图4A)。然而,与SDF和CB的联合干预导致香农和辛普森指数显著增加(p<0.05)。值得注意的是,这三项指标在HFD-CB和HFD-SDF组中并不显著。我们通过线性判别分析效应大小(LEfSe)比较了五组中的肠道微生物群,以确定不同组中的特定微生物群(图4B和C)。与NCD喂养的小鼠相比,HFD喂养的小鼠中梭状芽孢杆菌UCG-014的相对丰度显著降低 。然而,在HFD-SC组中,其他梭菌(Vadinb60梭菌)的相对丰度显著增加。这一发现表明,与SDF和CB的联合干预成功地促进了CB肠道定植。此外,乙酰化因子、气味细菌、粪便杆菌和肠杆菌(其成员已知为丁酸生产者)在HFD-SDF、HFD-CB或HFD-SC组中更为丰富。总之,这些结果表明,与SDF和CB的联合干预改善了HFD诱导的肠道微生物群紊乱,并增加了丁酸产生菌的数量。
3.5 SDF和CB联合干预改善HFD诱导的肠道粘膜屏障损伤
鉴于转录组测序的结果,我们接下来试图确定SD F和CB对肠粘膜的影响。肠绒毛不仅被认为是一种天然的保护屏障,而且是消化和营养吸收的主要场所。事实上,肠绒毛是脂质代谢和内毒素血症的关键因素。肠绒毛长度和表面的增加与营养素的吸收率有关。因此,肠绒毛长宽比通常用于评估肠道生长和功能。我们检查了小肠的组织形态学和长宽比,以确定CB和/或SDF治疗的效果。H&E染色显示,与N CD 喂养的小鼠相比,H FD 喂养的小鼠的绒毛长度和隐窝深度显著减少(图5A )。然而,在H FD 喂养的小鼠中,H FD -PBS组的肠绒毛长宽比显著低于其他组(图5B)。紧密连接蛋白是肠粘膜屏障的结构基础。对所有组的小肠组织进行蛋白质印迹,以检测紧密连接相关蛋白的表达(图5C )。蛋白质丰度图 5:SDF 和 CB 的联合干预改善了肠道粘膜的完整性。( A ) H & E 染色( 100× )小肠切片的代表性图片。( B )绒毛长宽比。( C )肠道紧密连接蛋白的代表性免疫印迹分析。( D )紧密连接蛋白丰度的定量分析。( E )L02 细胞中MitoSOX染色的代表性图像。数据以平均值±标准差表示,每组n=2–7。 a: p<0.05 ;与 NCD-PBS 相比, b:p<0.05 ;与 HFD-PBS 相比;c:p<0.05 ;与HFD-CB相比; d:p<0.05;与HFD-SDF相比; e:p<0.05 ;与HFD-SC相比。HFD-PBS组的claudin-4、claudin-1和ocludin显著低于其他四组(图5D )。此外,在HFD-SC 组中观察到紧密连接相关蛋白的最高表达。上述数据表明,与SDF和CB的联合干预改善了肠道粘膜屏障的结构和功能。
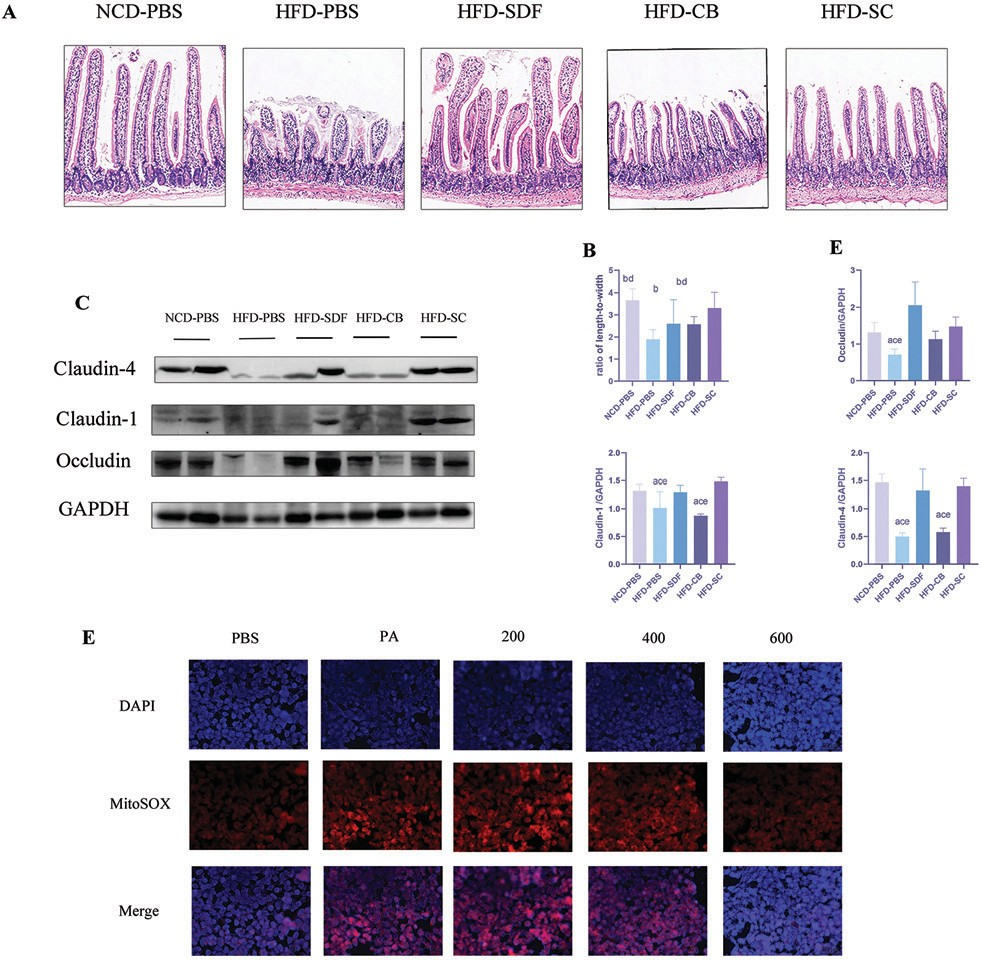
图 5:SDF 和 CB 的联合干预改善了肠道粘膜的完整性。(A)H&E染色(100×)小肠切片的代表性图片。(B)绒毛长宽比。(C)肠道紧密连接蛋白的代表性免疫印迹分析。(D)紧密连接蛋白丰度的定量分析。(E)L02 细胞中MitoSOX染色的代表性图像。数据以平均值 ± 标准差表示,每组n=2–7 。 a:p<0.05 ;与NCD-PBS 相比,b:p<0.05 ;与HFD-PBS 相比;c : p<0.05 ;与HFD-CB相比; d:p<0.05 ;与 HFD-SDF 相比; e:p<0.05 ;与HFD-SC相比。
3.6 SDF和CB联合干预改善HFD诱导的氧化应激和脂质代谢功能障碍
转录组测序结果表明,HFD通过氧化应激和脂质代谢功能障碍诱导MAFLD。为了研究联合干预是否通过调节与脂质代谢和抗氧化应激相关的基因表达来改善MAFLD ,我们重点研究了上述信号通路中的核心蛋白。乙酰辅酶A羧化酶(ACC )和ATP柠檬酸裂解酶(Acly)是催化脂肪酸合成的限速酶。 Nrf2是氧化应激信号的主要调节因子。结果表明,联合干预减少了H FD 诱导的Acly和ACC 磷酸化(图6A和B)。与其他脂质合成相关的蛋白质在HFD-SC组也明显减少,包括脂质1、脂肪酸合酶(FASN )、酰基辅酶A合成酶(ACSL1)和细胞质乙酰辅酶A合成酶(AceCS1)。然而,在HFD-CB和HFD-SDF组中,我们没有发现这些蛋白质有明显变化。除了高效的脂肪酸氧化外,氧化应激也是MAFLD的一个普遍特征。Nrf2是细胞抵抗氧化应激的中央防御蛋白。因此,我们探讨了联合干预在Nrf2表达中的作用。值得注意的是,结果表明,联合干预诱导了Nrf2、Keap1和超氧化物歧化酶(SOD)的表达(图6C和D )。丁酸盐是CB从SDF中提取的重要代谢产物。为了证实丁酸的影响,我们在L02细胞中使用PA诱导的MAFLD,但在Masson染色中未发现任何明显变化。接下来,我们分析了不同丁酸浓度下的蛋白质和活性氧水平。一直以来,丁酸降低了Acly和ACC磷酸化,激发了Nrf2活性并抑制了脂质合成(图6E和F)。从历史上看,减少活性氧是限制与氧化应激相关的细胞损伤的关键。丁酸剂量依赖性地降低L02细胞中的ROS水平(图5E)。总之,这些结果表明丁酸可能是改善MAFLD的关键。
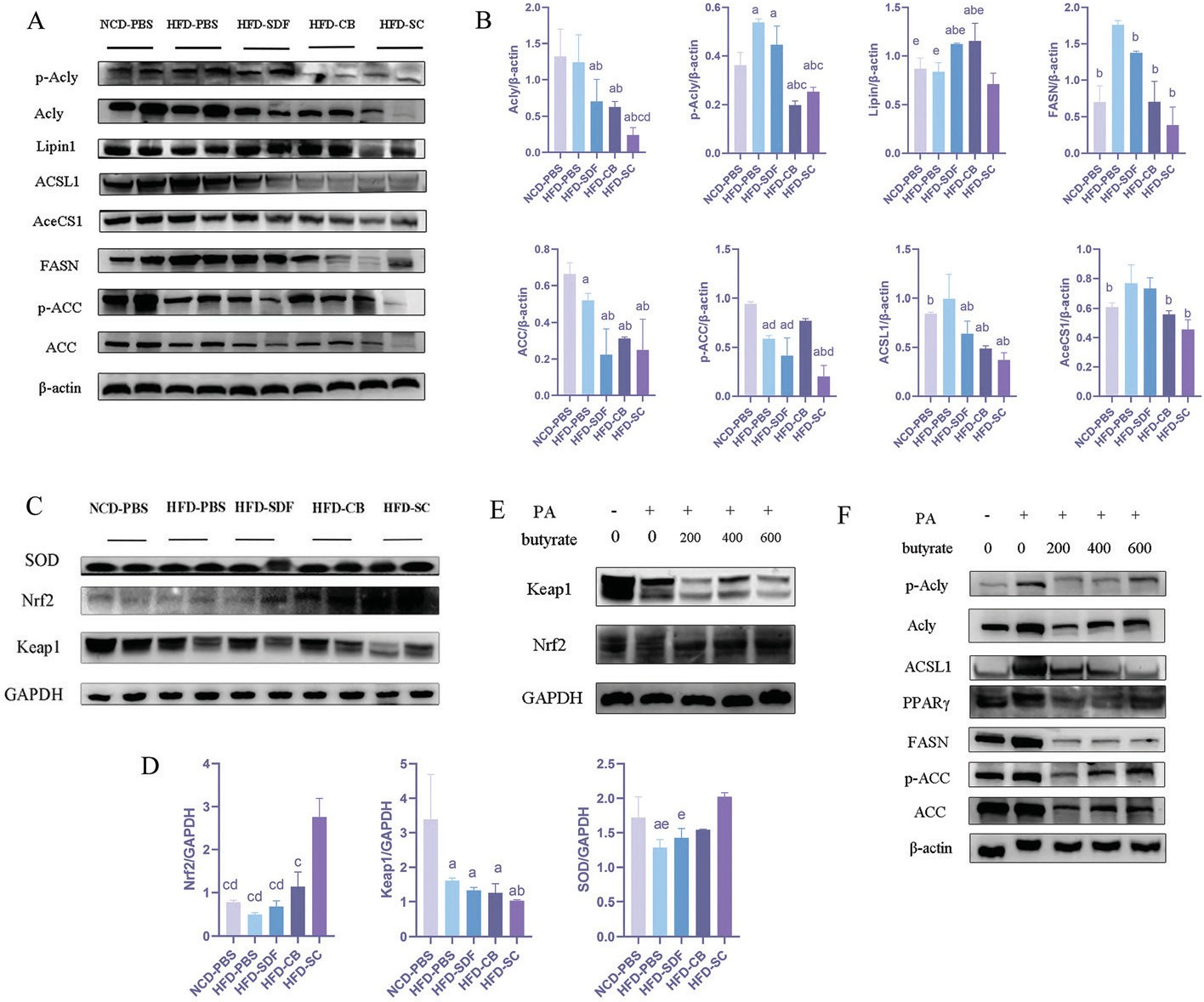
图6 SDF和CB的联合干预改善了脂质代谢和氧化应激。(A)肝脏中脂肪酸和脂质代谢蛋白的代表性免疫印迹分析。(B)脂肪酸和脂质代谢蛋白质丰度的定量分析。(C)肝脏中 Nrf2/Keap1 信号通路和 SOD 蛋白的代表性免疫印迹分析。(D)Nrf2/Keap1 信号通路和 SOD 蛋白丰度的定量分析。(E)L02 细胞中 Nrf2/Keap1 信号通路蛋白的代表性免疫印迹分析。(F) L02 细胞中脂肪酸和脂质代谢蛋白的代表性免疫印迹分析。数据以平均值 ± 标准差表示,每组 n=2 。 a:p<0.05;与 NCD-PBS相比,b:p<0.05;与 HFD-PBS相比;c:p<0.05 ;与HFD-CB相比;d:p<0.05;与HFD-SDF相比; e:p<0.05 ;与HFD-SC相比。
【讨论】
普遍认为,氧化应激、肠道微生物群紊乱和肝脏脂质代谢功能障碍在MAFLD发病机制中起关键作用。我们的结果表明,SDF和CB的影响存在个体差异。由于这些个体差异,很难评估SDF和CB在MAFLD中的作用。相反,SDF和CB的联合干预有效地保护了肠道完整性,降低了体重和血脂沉积和全身炎症,并增加微生物多样性。这些结果表明,通过针对肠道微生物群调节脂质代谢、氧化应激和炎症,与SDF和CB的联合干预对MAFLD 具有积极作用。MAFLD 患者通常伴有肠道微生物群失调和产生LPS的细菌增多。高脂肪饮食会导致血清LPS水平过高,进而通过减少紧密连接蛋白(claudin1、claudin4和ocludin)破坏肠粘膜完整性,并诱导肠上皮细胞凋亡。LPS-CD14复合物触发TLR4激活巨噬细胞,激活NF-κB信号通路和随后的N LRP3激活,诱导产生促炎细胞因子,如TNF-α、IFN -γ、IL-6和IL-1β。此外,100纳克/毫升LPS和20纳克/毫升IFN -γ刺激巨噬细胞M 1极化,然后瀑布效应和释放氧自由基。因此,LPS和M1巨噬细胞形成炎症正反馈环。CB通过发酵SDF产生丁酸和其他短链脂肪酸参与抗炎作用。同时,丁酸在线粒体中被有效吸收和氧化为乙酰辅酶a,作为肠上皮细胞的直接能源。此外,丁酸可以稳定缺氧诱导因子,进而增强上皮屏障功能。在线粒体基质中,脂肪酸β-氧化将脂肪酸转化为乙酰辅酶A。随后,乙酰辅酶A 通过三羧酸循环和氧化磷酸化氧化生成ATP。然而,ACC催化乙酰辅酶A转化为丙二酰辅酶A ,这是脂肪酸合成的第一步。ACC是AMPK的直接下游激酶。AMPK在ACC中Ser79处的磷酸化降低了ACC的酶活性。AMP/ATP比率的增加是AMPK激活的主要机制。乙酰辅酶A反式酯酶牺牲一个乙酰辅酶A来激活丁酸盐。值得注意的是,丁酸加速ATP消耗,并通过增加AMP/ATP的比率激活AMPK。这可能是联合干预导致AMPK激活和防止肝脏脂质积聚的原因。近年来,许多研究强调氧化应激促进NAFLD进展为NASH 。肠上皮细胞和肠道微生物群之间的相互作用诱导宿主细胞产生ROS。与LPS一起,ROS促进TLR4基因表达增加,进一步激活M1巨噬细胞诱导的炎症。此外,炎症促进持续氧化应激并导致线粒体功能障碍。在这种情况下,线粒体呼吸链疾病的特征是电子泄漏,导致活性氧的积累。以前的研究表明,肝损伤期间巨噬细胞的极化导致代谢重组,其特征是炎症小体激活和乳酸和活性氧的产生增加。抗氧化剂抑制TLR4/N F-κB信号通路的激活可以有效抑制炎症小体的激活,和M1巨噬细胞的极化。上文描述了HFD-PBS和HFD-SC组之间差异基因的GO分析。结果清楚地表明,许多与线粒体功能相关的基因显著富集,例如“呼吸链”、“线粒体内膜”、“氧化还原酶复合体/活性”、“过氧化物酶活性”和“抗氧化活性”。值得注意的是,联合干预可以激活Nrf2-Keap1信号通路,导致随后的抗氧化反应激活。Nrf2-Keap1信号通路是第二阶段抗氧化基因的主要激活因子,如SOD、CAT、GST、HO-1和NQO1.益生菌作为改善健康和预防疾病的手段疾病正面临殖民化的挑战。在我们的研究中,与SDF和CB的联合干预改善了MAFLD ,然而,与SDF或CB的单一干预并没有产生预期的结果。一个合理的解释是,SDF作为细菌发酵的基质有助于为炭黑的萌发和定植创造有利的环境。此外,丁酸还通过抑制炎症、维持肠粘膜完整性、和促进脂肪酸β氧化在改善MAFLD 中发挥重要作用。然而,一些研究报告称,低聚木糖和卟啉衍生低聚糖ides82可缓解小鼠MAFLD 和相关肠道微生物群失调。相反,VishalSingh等人发现,长期食用可溶性纤维菊糖、果胶或低聚果糖会导致小鼠肠道微生物群失调和肝脏疾病(胆汁淤积、肝脏炎症和肝癌)。可持续发展框架有多种,不同的可持续发展框架有不同的影响。是否有细菌专门利用SDF,单一或混合类型的SD F的影响以及不同类型的SD F对肠道微生物功能、宿主健康和代谢的影响仍不清楚。在新食品和药品的开发过程中,益生菌和SDF的选择是食品和医疗行业面临的挑战。
【结论】
我们已经确定,与SD F和C B的联合干预可以抑制M 1巨噬细胞的激活,防止H FD 诱导的慢性炎症,并促进脂质代谢。机制研究表明,共同干预改善了线粒体功能通过激活Acly/Nrf2/NF-κB信号通路来实现,该信号通路介导抗氧化能力和脂质代谢活性的增加,从而降低LPS、RO S和炎症反应的水平。与SD F和C B的联合干预为进一步发展微生物群靶向干预治疗和临床应用带来了巨大希望。